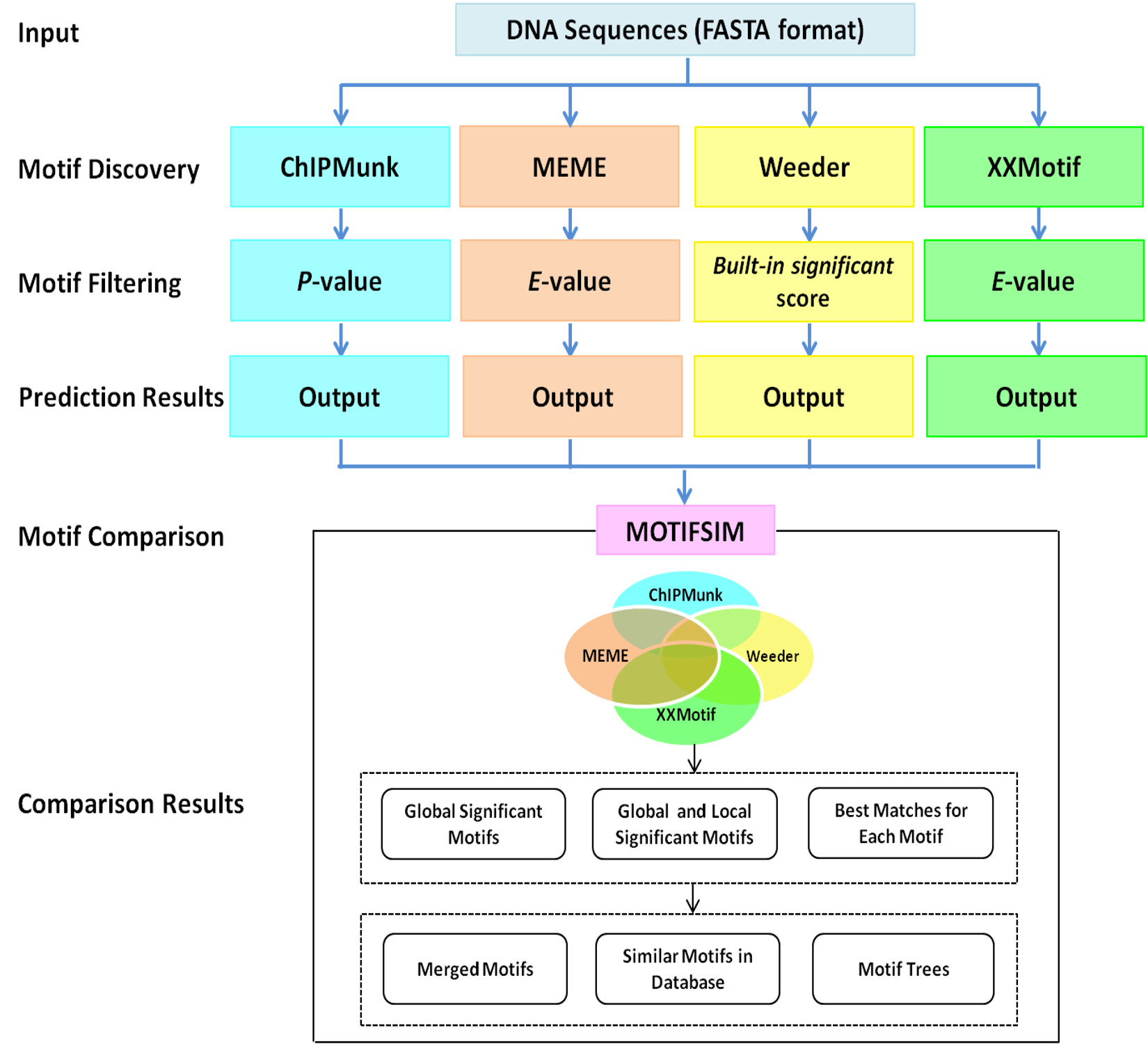
MODSIDE is a motif discovery pipeline and similarity detector. The pipeline integrated four de novo motif finders: ChIPMunk, MEME, Weeder, XXMotif, and a motif similarity detector MOTIFSIM. The results from motif finders are compared by using MOTIFSIM for identifying the global significant motifs, global and local significant motifs, and best matches for each motif. Users can obtain similar motifs in a reference database for the results. The motif trees can also be generated for the results. Similar motifs in the results can be merged to reduce the number of redundant motifs if needed. The predicted motifs from individual motif finders and the comparison results from MOTIFSIM can be viewed or downloaded. The program flow of the pipeline is illustrated below.
MODSIDE accepts DNA sequences in FASTA format with file extensions .fasta, .fa, and .txt. An example of input format is below.
>seq_0
TCTTTTTCCGTGCAAGATCAACGTTGAGCGCAAAATTGTTCGGAGACATTATCAACTCATTCATAGATCCATTAAACTGGAAAACACTATTTGTTATTTTTTG
>seq_1
CAATTCTACCGCATGCCTAACAAAATGATTTCCGATTTTTTCAATCTAACTCCGTCATTTCTTATCTGAGTTTTCCTCCCCCACCGGCGGTCTTTCGTCCGTG
>seq_2
GCCGGGCATACTCGGGATCTACCGTAGAATCCTATGCGTCACACATGAGGACAGGTTACGAACCGTCGAAGGCTTCGCGGCGTGTGTGATTAGTATTGTTTTG
>seq_3
CTGTCTAAACGATACAATCCTCGTTCCACCCACACCCATTCACCCGTCGGGCGAAATCCACCCTCGCGGCCTCGTTTAGAATACCAGGCAACGGCACCAGCAA
Input file can be uploaded or pasted on the browser with the format above.
It can be selected from the dropdown for running Weeder but not required. If an organism is unspecified, Weeder assumes it is Homo Sapiens.
At least two motif finders are required for running the pipeline. The tools and their versions are below.
| Tool | Version |
| ChIPMunk | v7 |
| MEME | 4.11.4_1 | Weeder | 1.4.2 | XXmotif | Current and only version (unspecified) | MOTIFSIM | 2.2 |
Number of Top Significant Motifs
This is a cutoff for number of top global significant motifs as well as number of top global and local significant motifs to be generated in the results. It is currently limited to ≤ 50. The default value is set to 5.
The number of best matches is the number of motifs that are most similar to motif i (i from 1 to m) in a combined motif list M in MOTIFSIM. It is used for selecting the number of most similar motifs to motif i and report them in MOTIFSIM's results. The best matched motifs are listed in the order of similarity with the most similar one is on the top of the list. The number of best matches is currently limited to ≤ 50. The default value is set to 5.
Currently, the cutoff values are ≥ 50%, ≥ 60%, ≥ 70%, ≥ 75%, ≥ 80%, ≥ 85%, and ≥ 90%. A value ≥ 75% indicates a match of 75% or greater between two motifs. We suggest to use a cutoff ≥ 75% as this value shows a good threshold in our case studies. If a higher cutoff value is used, fewer similar motifs are reported in the results. However, these motifs are more similar to the motif being compared.
Similar motifs in a reference database can be obtained for the global significant motifs, global and local significant motifs, as well as for every motif in the combined list M in MOTIFSIM. Currently, the pipeline supports Jaspar 2016 [1], Transfac [2] (public version), and UniPROBE [3] databases.
Motif trees can be generated for the global significant motifs as well as for the entire combined motif list M. The tree is built by using hclust function in R [4]. This function implemented the hierarchical clustering algorithm. MOTIFSIM generates the distance matrix for building the tree. This matrix contains the best similarity scores between motifs.
Similar motifs discovered in the results can be combined into new motifs. Two similar motifs can be combined into a new motif if the new motif is within the similarity threshold with both of its parents.
It can be (1) Global Significant Motifs Only or (2) All. The former generates only the global significant motifs in the results. The later generates all results and it requires longer time for processing large datasets.
The pipeline provides four options for MOTIFSIM's output file format: Text, HTML, PDF, and All formats. HTML, PDF, and All formats require longer time for processing large datasets.
Users can run test on sample data. The Use Sample Data button fills the required fields with sample data and the parameters below and submit it. The result page appears when the submitted job is completed.
| Parameter | Value |
| Input (sample data) |
>seq_0 |
| Organism | Homo sapiens |
| Motif Finder |
ChIPMunk Weeder |
| Number of top significant motifs | 3 |
| Number of best matches | 3 |
| Cutoff for similarity | 75% |
| Databse match | No selection |
| Motif tree | No |
| Combine similar motifs | No |
| Output file type | All |
| Output file format | Text |
Optional. If it is provided, users will be notified when a submitted job is completed and ready for viewing and downloading.
The results from individual motif finder are available for viewing and downloading. The comparison results from MOTIFSIM can be viewed and downloaded in HTML, PDF, Text, and all three formats. For more information on comparison results, please see MOTIFSIM's user manual.
References
| 1. | Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B: JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Research 2004, 32:D91-D94. |
| 2. | Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Munch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E: TRANSFAC®: transcriptional regulation, from patterns to profiles. Nucleic Acids Research 2003, 31(1):374-8. |
| 3. | Newburger N and Bulyk M. UniPROBE: an online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Research 2009, 37:D77-D82. |
| 4. | R Core Team (2016). R: A language and environment for statistical computing. R Foundation for Statistical Computing , Vienna, Austria. URL https://www.R-project.org/. |